One film-coated tablet contains 150 mg Erlotinib (as Erlotinib hydrochloride).
Excipients/Inactive Ingredients: Lactose Monohydrate; Cellulose, Microcrystalline; Sodium Starch Glycolate; Magnesium Stearate; Isopropyl Alcohol; Opadry 200 white.
Pharmacotherapeutic Group: Antineoplastic agent protein kinase inhibitor. ATC Code: L01XE03.
Pharmacology: Pharmacodynamics: Mechanism of Action: Erlotinib is an epidermal growth factor receptor/human epidermal growth factor receptor type 1 (EGFR also known as HER1) tyrosine kinase inhibitor. Erlotinib potently inhibits the intracellular phosphorylation of EGFR. EGFR is expressed on the cell surface of normal cells and cancer cells. In non-clinical models, inhibition of EGFR phosphotyrosine results in cell stasis and/or death.
EGFR mutations may lead to constitutive activation of anti-apoptotic and proliferation signaling pathways. The potent effectiveness of Erlotinib in blocking EGFR-mediated signalling in these EGFR mutation positive tumours is attributed to the tight binding of Erlotinib to the ATP-binding site in the mutated kinase domain of the EGFR. Due to the blocking of downstream-signaling, the proliferation of cells is stopped, and cell death is induced through the intrinsic apoptotic pathway. Tumour regression is observed in mouse models of enforced expression of these EGFR activating mutations.
Pharmacokinetics: Absorption: After oral administration, Erlotinib peak plasma levels are obtained in approximately 4 hours after oral dosing. A study in normal healthy volunteers provided an estimate of the absolute bioavailability of 59%. The exposure after an oral dose may be increased by food.
Distribution: Erlotinib has a mean apparent volume of distribution of 232 l and distributes into tumour tissue of humans.
In a study of 4 patients (3 with non-small cell lung cancer [NSCLC], and 1 with laryngeal cancer) receiving 150 mg daily oral doses of Erlotinib, tumour samples from surgical excisions on Day 9 of treatment revealed tumour concentrations of Erlotinib that averaged 1185 ng/g of tissue. This corresponded to an overall average of 63% (range 5-161%) of the steady state observed peak plasma concentrations. The primary active metabolites were present in tumour at concentrations averaging 160 ng/g tissue, which corresponded to an overall average of 113% (range 88-130%) of the observed steady state peak plasma concentrations. Plasma protein binding is approximately 95%.
Erlotinib binds to serum albumin and alpha-1 acid glycoprotein (AAG).
Biotransformation: Erlotinib is metabolised in the liver by the hepatic cytochromes in humans, primarily CYP3A4 and to a lesser extent by CYP1A2. Extrahepatic metabolism by CYP3A4 in intestine, CYP1A1 in lung, and 1B1 in tumour tissue potentially contribute to the metabolic clearance of Erlotinib.
There are three main metabolic pathways identified: 1) O-demethylation of either side chain or both, followed by oxidation to the carboxylic acids; 2) oxidation of the acetylene moiety followed by hydrolysis to the aryl carboxylic acid; and 3) aromatic hydroxylation of the phenyl-acetylene moiety. The primary metabolites OSI-420 and OSI-413 of Erlotinib produced by O-demethylation of either side chain have comparable potency to Erlotinib in non-clinical in vitro assays and in vivo tumour models. They are present in plasma at levels that are <10% of Erlotinib and display similar pharmacokinetics as Erlotinib.
Elimination: Erlotinib is excreted predominantly as metabolites via the faeces (>90%) with renal elimination accounting for only a small amount (approximately 9%) of an oral dose. Less than 2% of the orally administered dose is excreted as parent substance. A population pharmacokinetic analysis in 591 patients receiving single agent Erlotinib shows a mean apparent clearance of 4.47 l/hour with a median half-life of 36.2 hours. Therefore, the time to reach steady state plasma concentration would be expected to occur in approximately 7-8 days.
Pharmacokinetics in special population: Based on population pharmacokinetic analysis, no clinically significant relationship between predicted apparent clearance and patient age, bodyweight, gender and ethnicity were observed. Patient factors, which correlated with Erlotinib pharmacokinetics, were serum total bilirubin, AAG and current smoking. Increased serum concentrations of total bilirubin and AAG concentrations were associated with a reduced Erlotinib clearance. The clinical relevance of these differences is unclear. However, smokers had an increased rate of Erlotinib clearance. This was confirmed in a pharmacokinetic study in non-smoking and currently cigarette smoking healthy subjects receiving a single oral dose of 150 mg Erlotinib. The geometric mean of the Cmax was 1056 ng/mL in the non-smokers and 689 ng/mL in the smokers with a mean ratio for smokers to non-smokers of 65.2% (95% CI: 44.3 to 95.9, p = 0.031). The geometric mean of the AUC0-inf was 18726 ng•h/mL in the non-smokers and 6718 ng•h/mL in the smokers with a mean ratio of 35.9% (95% CI: 23.7 to 54.3, p < 0.0001). The geometric mean of the C24h was 288 ng/mL in the non-smokers and 34.8 ng/mL in the smokers with a mean ratio of 12.1% (95% CI: 4.82 to 30.2, p = 0.0001).
In the pivotal Phase III NSCLC trial, current smokers achieved Erlotinib steady state trough plasma concentration of 0.65 μg/mL (n=16) which was approximately 2-fold less than the former smokers or patients who had never smoked (1.28 μg/mL, n=108). This effect was accompanied by a 24% increase in apparent Erlotinib plasma clearance. In a phase I dose escalation study in NSCLC patients who were current smokers, pharmacokinetic analyses at steady-state indicated a dose proportional increase in Erlotinib exposure when the Erlotinib dose was increased from 150 mg to the maximum tolerated dose of 300 mg. Steady-state trough plasma concentrations at a 300 mg dose in current smokers in this study was 1.22 μg/mL (n=17).
Based on the results of pharmacokinetic studies, current smokers should be advised to stop smoking while taking Erlotinib, as plasma concentrations could be reduced otherwise.
Based on population pharmacokinetic analysis, the presence of an opioid appeared to increase exposure by about 11%.
A second population pharmacokinetic analysis was conducted that incorporated Erlotinib data from 204 pancreatic cancer patients who received Erlotinib plus gemcitabine. This analysis demonstrated that covariants affecting Erlotinib clearance in patients from the pancreatic study were very similar to those seen in the prior single agent pharmacokinetic analysis. No new covariate effects were identified. Co-administration of gemcitabine had no effect on Erlotinib plasma clearance.
Paediatric population: There have been no specific studies in paediatric patients.
Elderly population: There have been no specific studies in elderly patients.
Hepatic impairment: Erlotinib is primarily cleared by the liver. In patients with solid tumours and with moderately impaired hepatic function (Child-Pugh score 7-9), geometric mean Erlotinib AUC0-t and Cmax was 27000 ng•h/mL and 805 ng/mL, respectively, as compared to 29300 ng•h/mL and 1090 ng/mL in patients with adequate hepatic function including patients with primary liver cancer or hepatic metastases. Although the Cmax was statistically significant lower in moderately hepatic impaired patients, this difference is not considered clinically relevant. No data are available regarding the influence of severe hepatic dysfunction on the pharmacokinetics of Erlotinib. In population pharmacokinetic analysis, increased serum concentrations of total bilirubin were associated with a slower rate of Erlotinib clearance.
Renal impairment: Erlotinib and its metabolites are not significantly excreted by the kidney, as less than 9% of a single dose is excreted in the urine. In population pharmacokinetic analysis, no clinically significant relationship was observed between Erlotinib clearance and creatinine clearance, but there are no data available for patients with creatinine clearance <15 ml/min.
Toxicology: Preclinical data: Chronic dosing effects observed in at least one animal species or study included effects on the cornea (atrophy, ulceration), skin (follicular degeneration and inflammation, redness, and alopecia), ovary (atrophy), liver (liver necrosis), kidney (renal papillary necrosis and tubular dilatation), and gastrointestinal tract (delayed gastric emptying and diarrhoea). Red blood cell parameters were decreased and white blood cells, primarily neutrophils, were increased. There were treatment-related increases in ALT, AST and bilirubin. These findings were observed at exposures well below clinically relevant exposures.
Based on the mode of action, Erlotinib has the potential to be a teratogen. Data from reproductive toxicology tests in rats and rabbits at doses near the maximum tolerated dose and/or maternally toxic doses showed reproductive (embryotoxicity in rats, embryo resorption and foetotoxicity in rabbits) and developmental (decrease in pup growth and survival in rats) toxicity, but was not teratogenic and did not impair fertility. These findings were observed at clinically relevant exposures.
Erlotinib tested negative in conventional genotoxicity studies. Two-year carcinogenicity studies with Erlotinib conducted in rats and mice were negative up to exposures exceeding human therapeutic exposure (up to 2-fold and 10-fold higher, respectively, based on Cmax and/or AUC).
A mild phototoxic skin reaction was observed in rats after UV irradiation.
Non-Small Cell Lung Cancer (NSCLC): Erlotinib Sandoz is indicated for the treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of at least one prior chemotherapy regimen.
Pancreatic cancer: Erlotinib Sandoz in combination with gemcitabine is indicated for the first-line treatment of patients with locally advanced, unresectable or metastatic pancreatic cancer.
Erlotinib Sandoz treatment should be supervised by a physician experienced in the use of anti-cancer therapies.
Patients with Non-Small Cell Lung Cancer: EGFR mutation testing should be performed prior to initiation of Erlotinib Sandoz therapy in chemo-naïve patients with advanced or metastatic NSCLC.
The recommended daily dose of Erlotinib Sandoz is 150 mg taken at least one hour before or two hours after the ingestion of food.
Patients with pancreatic cancer: The recommended daily dose of Erlotinib Sandoz is 100 mg taken at least one hour before or two hours after the ingestion of food, in combination with gemcitabine (see the summary of product characteristics of gemcitabine for the pancreatic cancer indication). In patients who do not develop rash within the first 4 - 8 weeks of treatment, further Erlotinib Sandoz treatment should be re-assessed.
When dose adjustment is necessary, the dose should be reduced in 50 mg steps (see Precautions). Erlotinib Sandoz is available in strengths of 25 mg, 100 mg and 150 mg. Concomitant use of CYP3A4 substrates and modulators may require dose adjustment (see Interactions).
Patients with hepatic impairment: Erlotinib Sandoz is eliminated by hepatic metabolism and biliary excretion. Although Erlotinib exposure was similar in patients with moderately impaired hepatic function (Child-Pugh score 7-9) compared with patients with adequate hepatic function, caution should be used when administering Erlotinib Sandoz to patients with hepatic impairment. Dose reduction or interruption of Erlotinib Sandoz should be considered if severe adverse reactions occur. The safety and efficacy of Erlotinib has not been studied in patients with severe hepatic dysfunction (AST/SGOT and ALT/SGPT> 5 x ULN). Use of Erlotinib Sandoz in patients with severe hepatic dysfunction is not recommended.
Patients with renal impairment: The safety and efficacy of Erlotinib has not been studied in patients with renal impairment (serum creatinine concentration >1.5 times the upper normal limit). Based on pharmacokinetic data no dose adjustments appear necessary in patients with mild or moderate renal impairment (see Pharmacology: Pharmacokinetics under Actions). Use of Erlotinib in patients with severe renal impairment is not recommended.
Paediatric population: The safety and efficacy of Erlotinib in patients under the age of 18 years has not been established. Use of Erlotinib Sandoz in paediatric patients is not recommended.
Smokers: Cigarette smoking has been shown to reduce Erlotinib exposure by 50-60%. The maximum tolerated dose of Erlotinib Sandoz in NSCLC patients who currently smoke cigarettes was 300 mg. Efficacy and long term safety of a dose higher than the recommended starting doses have not been established in patients who continue to smoke cigarettes (see Interactions and Pharmacology: Pharmacokinetics under Actions). Therefore, current smokers should be advised to stop smoking, as plasma concentrations of Erlotinib in smokers as compared to non-smokers are reduced.
Symptoms: Single oral doses of Erlotinib up to 1000 mg Erlotinib in healthy subjects, and up to 1600 mg in cancer patients have been tolerated. Repeated twice daily doses of 200 mg in healthy subjects were poorly tolerated after only a few days of dosing. Based on the data from these studies, severe adverse reactions such as diarrhoea, rash and possibly increased activity of liver aminotransferases may occur above the recommended dose.
Management: In case of suspected overdose, Erlotinib should be withheld and symptomatic treatment initiated.
Hypersensitivity to Erlotinib or to any of the excipients listed in Description.
Assessment of EGFR mutation status: When assessing the EGFR mutation status of a patient, it is important that a well- validated and robust methodology is chosen to avoid false negative or false positive determinations.
Smokers: Current smokers should be advised to stop smoking, as plasma concentrations of Erlotinib in smokers as compared to non-smokers are reduced. The degree of reduction is likely to be clinically significant (see Interactions).
Interstitial Lung Disease: Cases of interstitial lung disease (ILD)-like events, including fatalities, have been reported uncommonly in patients receiving Erlotinib for treatment of non-small cell lung cancer (NSCLC), pancreatic cancer or other advanced solid tumours. In the pivotal study BR.21 in NSCLC, the incidence of ILD (0.8%) was the same in both the placebo and Erlotinib groups. In the pancreatic cancer study in combination with gemcitabine, the incidence of ILD-like events was 2.5% in the Erlotinib plus gemcitabine group versus 0.4% in the placebo plus gemcitabine treated group. The overall incidence in Erlotinib -treated patients from all studies (including uncontrolled studies and studies with concurrent chemotherapy) is approximately 0.6% compared to 0.2% in patients on placebo. Reported diagnoses in patients suspected of having ILD-like events included pneumonitis, radiation pneumonitis, hypersensitivity pneumonitis, interstitial pneumonia, interstitial lung disease, obliterative bronchiolitis, pulmonary fibrosis, Acute Respiratory Distress Syndrome (ARDS), alveolitis, and lung infiltration. Symptoms started from a few days to several months after initiating Erlotinib therapy. Confounding or contributing factors such as concomitant or prior chemotherapy, prior radiotherapy, pre-existing parenchymal lung disease, metastatic lung disease, or pulmonary infections were frequent. A higher incidence of ILD (approximately 5% with a mortality rate of 1.5%) is seen among patients with Japanese origin.
In patients who develop acute onset of new and/or progressive unexplained pulmonary symptoms such as dyspnea, cough and fever, Erlotinib therapy should be interrupted pending diagnostic evaluation.
Patients treated concurrently with Erlotinib and gemcitabine should be monitored carefully for the possibility to develop ILD-like toxicity. If ILD is diagnosed, Erlotinib should be discontinued and appropriate treatment initiated as necessary (see Adverse Reactions).
Diarrhoea, dehydration, electrolyte imbalance and renal failure: Diarrhoea (including very rare cases with a fatal outcome) has occurred in approximately 50% of patients on Erlotinib and moderate or severe diarrhoea should be treated with e.g. loperamide. In some cases dose reduction may be necessary. In the clinical studies doses were reduced by 50 mg steps.
Dose reductions by 25 mg steps have not been investigated. In the event of severe or persistent diarrhoea, nausea, anorexia, or vomiting associated with dehydration, Erlotinib therapy should be interrupted and appropriate measures should be taken to treat the dehydration (see Adverse Reactions). There have been rare reports of hypokalaemia and renal failure (including fatalities). Some cases were secondary to severe dehydration due to diarrhoea, vomiting and/or anorexia, while others were confounded by concomitant chemotherapy. In more severe or persistent cases of diarrhoea, or cases leading to dehydration, particularly in groups of patients with aggravating risk factors (especially concomitant chemotherapy and other medications, symptoms or diseases or other predisposing conditions including advanced age), Erlotinib therapy should be interrupted and appropriate measures should be taken to intensively rehydrate the patients intravenously. In addition, renal function and serum electrolytes including potassium should be monitored in patients at risk of dehydration.
Hepatitis, hepatic failure: Rare cases of hepatic failure (including fatalities) have been reported during use of Erlotinib.
Confounding factors have included pre-existing liver disease or concomitant hepatotoxic medications. Therefore, in such patients, periodic liver function testing should be considered. Erlotinib dosing should be interrupted if changes in liver function are severe (see Adverse Reactions). Erlotinib is not recommended for use in patients with severe hepatic dysfunction.
Gastrointestinal perforation: Patients receiving Erlotinib are at increased risk of developing gastrointestinal perforation, which was observed uncommonly (including some cases with a fatal outcome). Patients receiving concomitant anti-angiogenic agents, corticosteroids, NSAIDs, and/or taxane based chemotherapy, or who have prior history of peptic ulceration or diverticular disease are at increased risk. Erlotinib should be permanently discontinued in patients who develop gastrointestinal perforation (see Adverse Reactions).
Bullous and exfoliative skin disorders: Bullous, blistering and exfoliative skin conditions have been reported, including very rare cases suggestive of Stevens-Johnson syndrome/Toxic epidermal necrolysis, which in some cases were fatal (see Adverse Reactions). Erlotinib treatment should be interrupted or discontinued if the patient develops severe bullous, blistering or exfoliating conditions. Patients with bullous and exfoliative skin disorders should be tested for skin infection and treated according to local management guidelines.
Ocular disorders: Patients presenting with signs and symptoms suggestive of keratitis such as acute or worsening: eye inflammation, lacrimation, light sensitivity, blurred vision, eye pain and/or red eye should be referred promptly to an ophthalmology specialist. If a diagnosis of ulcerative keratitis is confirmed, treatment with Erlotinib should be interrupted or discontinued. If keratitis is diagnosed, the benefits and risks of continuing treatment should be carefully considered. Erlotinib should be used with caution in patients with a history of keratitis, ulcerative keratitis or severe dry eye. Contact lens use is also a risk factor for keratitis and ulceration. Very rare cases of corneal perforation or ulceration have been reported during use of Erlotinib (see Adverse Reactions).
Interactions with other medicinal products: Potent inducers of CYP3A4 may reduce the efficacy of Erlotinib whereas potent inhibitors of CYP3A4 may lead to increased toxicity. Concomitant treatment with these types of agents should be avoided (see Interactions).
Other forms of interactions: Erlotinib is characterised by a decrease in solubility at pH above 5. Medicinal products that alter the pH of the upper Gastro-Intestinal (GI) tract, like proton pump inhibitors, H2 antagonists and antacids, may alter the solubility of Erlotinib and hence its bioavailability. Increasing the dose of Erlotinib when co-administered with such agents is not likely to compensate for the loss of exposure. Combination of Erlotinib with proton pump inhibitors should be avoided. The effects of concomitant administration of Erlotinib with H2 antagonists and antacids are unknown; however, reduced bioavailability is likely.
Therefore, concomitant administration of these combinations should be avoided (see Interactions). If the use of antacids is considered necessary during treatment with Erlotinib, they should be taken at least 4 hours before or 2 hours after the daily dose of Erlotinib.
The tablets contain lactose and should not be administered to patients with rare hereditary problems of galactose intolerance, Lapp lactase deficiency or glucose-galactose malabsorption.
Effects on ability to drive and use machines: No studies on the effects on the ability to drive and use machines have been performed; however Erlotinib is not associated with impairment of mental ability.
Pregnancy: There are no adequate data for the use of Erlotinib in pregnant women. Studies in animals have shown no evidence of teratogenicity or abnormal parturition. However, an adverse effect on the pregnancy cannot be excluded as rat and rabbit studies have shown increased embryo/foetal lethality, (see Pharmacology: Toxicology: Preclinical data under Actions). The potential risk for humans is unknown.
Women of child bearing potential: Women of childbearing potential must be advised to avoid pregnancy while on Erlotinib. Adequate contraceptive methods should be used during therapy, and for at least 2 weeks after completing therapy. Treatment should only be continued in pregnant women if the potential benefit to the mother outweighs the risk to the foetus.
Breast-feeding: It is not known whether Erlotinib is excreted in human milk. Because of the potential harm to the infant, mothers should be advised against breast-feeding while receiving Erlotinib.
Fertility: Studies in animals have shown no evidence of impaired fertility. However, an adverse effect on the fertility cannot be excluded as animal studies have shown effects on reproductive parameters (see Pharmacology: Toxicology: Preclinical data under Actions). The potential risk for humans is unknown.
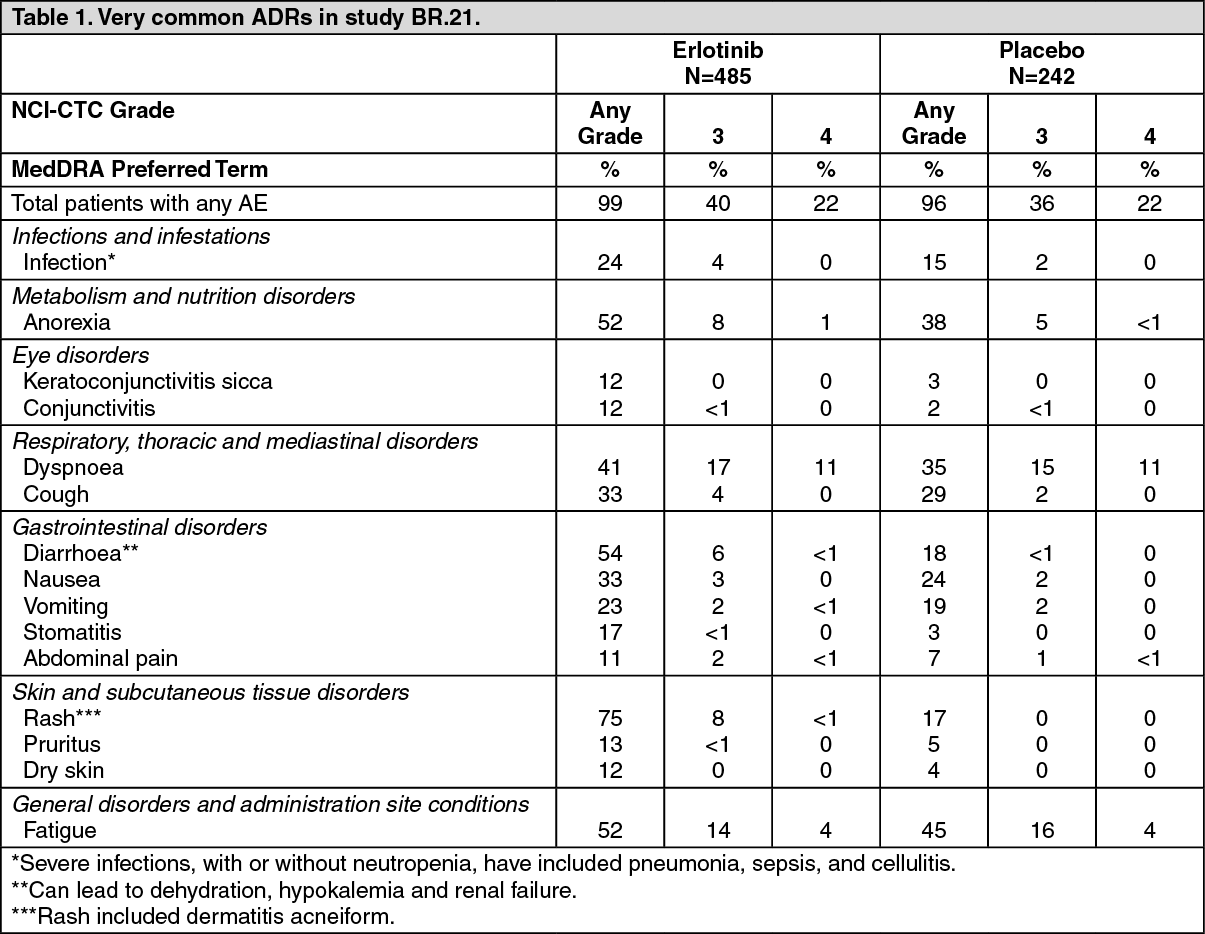
Non-small cell lung cancer (Erlotinib administered as monotherapy): In a randomized double-blind study (BR.21; Erlotinib administered as second line therapy), rash (75%) and diarrhoea (54%) were the most commonly reported adverse drug reactions (ADRs). Most were Grade 1/2 in severity and manageable without intervention. Grade 3/4 rash and diarrhoea occurred in 9% and 6%, respectively in Erlotinib-treated patients and each resulted in study discontinuation in 1% of patients. Dose reduction for rash and diarrhoea was needed in 6% and 1% of patients, respectively.
In study BR.21, the median time to onset of rash was 8 days, and the median time to onset of diarrhea was 12 days.
In general, rash manifests as a mild or moderate erythematous and papulopustular rash, which may occur or worsen in sun exposed areas. For patients who are exposed to sun, protective clothing, and/or use of sun screen (e.g. mineral-containing) may be advisable.
Adverse reactions occurring more frequently (≥3%) in Erlotinib -treated patients than in the placebo group in the pivotal study BR.21, and in at least 10% of patients in the Erlotinib group, are summarized by National Cancer Institute-Common Toxicity Criteria (NCI-CTC) Grade in Table 1. (See Table 1.)
The following terms are used to rank the undesirable effects by frequency: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000) including isolated reports.
Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
In another double-blind, randomized, placebo-controlled Phase III study BO18192 (SATURN); Erlotinib was administered as maintenance after first-line chemotherapy. SATURN was conducted in 889 patients with advanced, recurrent or metastatic NSCLC following first-line standard platinumbased chemotherapy, no new safety signals were identified.
The most frequent ADRs seen in patients treated with Erlotinib in study BO18192 were rash and diarrhoea (any Grade 49% and 20%, respectively), most were Grade 1/2 in severity and manageable without intervention. Grade 3 rash and diarrhoea occurred in 6% and 2% of patients, respectively. No Grade 4 rash or diarrhoea was observed. Rash and diarrhoea resulted in discontinuation of Erlotinib in 1% and <1% of patients, respectively. Dose modifications (interruptions or reductions) for rash and diarrhoea were needed in 8.3% and 3% of patients, respectively.
In an open-label, randomized phase III study, ML20650 conducted in 154 patients, the safety of Erlotinib for first-line treatment of NSCLC patients with EGFR activating mutations was assessed in 75 patients; no new safety signals were observed in these patients.
The most frequent ADRs seen in patients treated with Erlotinib in study ML20650 were rash and diarrhoea (any Grade 80% and 57%, respectively), most were Grade 1/2 in severity and manageable without intervention. Grade 3 rash and diarrhoea occurred in 9% and 4% of patients, respectively. No Grade 4 rash or diarrhoea was observed. Both rash and diarrhoea resulted in discontinuation of Erlotinib in 1% of patients. Dose modifications (interruptions or reductions) for rash and diarrhea were needed in 11% and 7% of patients, respectively.
Pancreatic cancer (Erlotinib administered concurrently with gemcitabine): The most common adverse reactions in pivotal study PA.3 in pancreatic cancer patients receiving Erlotinib 100 mg plus gemcitabine were fatigue, rash and diarrhoea. In the Erlotinib plus gemcitabine arm, Grade 3/4 rash and diarrhoea were each reported in 5% of patients. The median time to onset of rash and diarrhoea was 10 days and 15 days, respectively. Rash and diarrhoea each resulted in dose reductions in 2% of patients, and resulted in study discontinuation in up to 1% of patients receiving Erlotinib plus gemcitabine.
Adverse reactions occurring more frequently (≥3%) in Erlotinib 100 mg plus gemcitabine-treated patients than in the placebo plus gemcitabine group in the pivotal study PA.3, and in at least 10% of patients in the Erlotinib 100 mg plus gemcitabine group, are summarised by National Cancer Institute- Common Toxicity Criteria (NCI- CTC) Grade in Table 2. (See Table 2).
The following terms are used to rank the undesirable effects by frequency: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000) including isolated reports.
Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
Click on icon to see table/diagram/image
Other Observations: Safety evaluation of Erlotinib is based on the data from more than 1200 patients treated with at least one 150 mg dose of Erlotinib monotherapy and more than 300 patients who received Erlotinib 100 or 150 mg in combination with gemcitabine.
The following adverse reactions have been observed in patients who received Erlotinib administered as single agent and patients who received Erlotinib concurrently with chemotherapy.
Very common ADRs from the BR 21 and PA 3 studies are presented in Tables 1 and 2, other ADRs including those from other studies are summarized in Table 3. (See Table 3.)
Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
Click on icon to see table/diagram/image
Interaction studies have only been performed in adults.
Erlotinib and other CYP substrates: Erlotinib is a potent inhibitor of CYP1A1, and a moderate inhibitor of CYP3A4 and CYP2C8, as well as a strong inhibitor of glucuronidation by UGT1A1 in vitro.
The physiological relevance of the strong inhibition of CYP1A1 is unknown due to the very limited expression of CYP1A1 in human tissues.
When Erlotinib was co-administered with ciprofloxacin, a moderate CYP1A2 inhibitor, the Erlotinib exposure [AUC] increased significantly by 39%, while no statistically significant change in Cmax was found. Similarly, the exposure to the active metabolite increased by about 60% and 48% for AUC and Cmax, respectively. The clinical relevance of this increase has not been established. Caution should be exercised when ciprofloxacin or potent CYP1A2 inhibitors (e.g. fluvoxamine) are combined with Erlotinib. If adverse reactions related to Erlotinib are observed, the dose of Erlotinib may be reduced.
Pre-treatment or co-administration of Erlotinib did not alter the clearance of the prototypical CYP3A4 substrates, midazolam and erythromycin, but did appear to decrease the oral bioavailability of midazolam by up to 24%. In another clinical study, Erlotinib was shown not to affect pharmacokinetics of the concomitantly administered CYP3A4/2C8 substrate paclitaxel. Significant interactions with the clearance of other CYP3A4 substrates are therefore unlikely.
The inhibition of glucuronidation may cause interactions with medicinal products which are substrates of UGT1A1 and exclusively cleared by this pathway. Patients with low expression levels of UGT1A1 or genetic glucuronidation disorders (e.g. Gilbert's disease) may exhibit increased serum concentrations of bilirubin and must be treated with caution.
Erlotinib is metabolised in the liver by the hepatic cytochromes in humans, primarily CYP3A4 and to a lesser extent by CYP1A2. Extrahepatic metabolism by CYP3A4 in intestine, CYP1A1 in lung, and CYP1B1 in tumour tissue also potentially contribute to the metabolic clearance of Erlotinib. Potential interactions may occur with active substances which are metabolised by, or are inhibitors or inducers of, these enzymes.
Potent inhibitors of CYP3A4 activity decrease Erlotinib metabolism and increase Erlotinib plasma concentrations. In a clinical study, the concomitant use of Erlotinib with ketoconazole (200 mg orally twice daily for 5 days), a potent CYP3A4 inhibitor, resulted in an increase of Erlotinib exposure (86% of AUC and 69% of Cmax). Therefore, caution should be used when Erlotinib is combined with a potent CYP3A4 inhibitor, e.g. azole antifungals (i.e. ketoconazole, itraconazole, voriconazole), protease inhibitors, erythromycin or clarithromycin. If necessary the dose of Erlotinib should be reduced, particularly if toxicity is observed.
Potent inducers of CYP3A4 activity increase Erlotinib metabolism and significantly decrease Erlotinib plasma concentrations. In a clinical study, the concomitant use of Erlotinib and rifampicin (600 mg orally once daily for 7 days), a potent CYP3A4 inducer, resulted in a 69% decrease in the median Erlotinib AUC. Co-administration of rifampicin with a single 450 mg dose of Erlotinib resulted in a mean Erlotinib exposure (AUC) of 57.5% of that after a single 150 mg Erlotinib dose in the absence of rifampicin treatment. Co-administration of Erlotinib with CYP3A4 inducers should therefore be avoided. For patients who require concomitant treatment with Erlotinib and a potent CYP3A4 inducer such as rifampicin an increase in dose to 300 mg should be considered while their safety (including renal and liver functions and serum electrolytes) is closely monitored, and if well tolerated for more than 2 weeks, further increase to 450 mg could be considered with close safety monitoring. Reduced exposure may also occur with other inducers e.g. phenytoin, carbamazepine, barbiturates or St. John's Wort (Hypericum perforatum). Caution should be observed when these active substances are combined with Erlotinib. Alternate treatments lacking potent CYP3A4 inducing activity should be considered when possible.
Erlotinib and coumarin-derived anticoagulants: Interaction with coumarin-derived anticoagulants including warfarin leading to increased International Normalized Ratio (INR) and bleeding events, which in some cases were fatal, have been reported in patients receiving Erlotinib. Patients taking coumarin-derived anticoagulants should be monitored regularly for any changes in prothrombin time or INR.
Erlotinib and statins: The combination of Erlotinib and a statin may increase the potential for statin- induced myopathy, including rhabdomyolysis, which was observed rarely.
Erlotinib and smokers: Results of a pharmacokinetic interaction study indicated a significant 2.8-, 1.5- and 9- fold reduced AUCinf, Cmax and plasma concentration at 24 hours, respectively, after administration of Erlotinib in smokers as compared to non-smokers (see Pharmacology: Pharmacokinetics under Actions). Therefore, patients who are still smoking should be encouraged to stop smoking as early as possible before initiation of treatment with Erlotinib, as plasma Erlotinib concentrations are reduced otherwise. The clinical effect of the decreased exposure has not been formally assessed but it is likely to be clinically significant.
Erlotinib and P-glycoprotein inhibitors: Erlotinib is a substrate for the P-glycoprotein active substance transporter. Concomitant administration of inhibitors of Pgp, e.g. cyclosporine and verapamil, may lead to altered distribution and/or altered elimination of Erlotinib. The consequences of this interaction for e.g. CNS toxicity have not been established. Caution should be exercised in such situations.
Erlotinib and medicinal products altering pH: Erlotinib is characterised by a decrease in solubility at pH above 5. Medicinal products that alter the pH of the upper Gastro-Intestinal (GI) tract may alter the solubility of Erlotinib and hence its bioavailability. Co-administration of Erlotinib with omeprazole, a proton pump inhibitor (PPI), decreased the Erlotinib exposure [AUC] and maximum concentration [Cmax] by 46% and 61%, respectively. There was no change to Tmax or half-life. Concomitant administration of Erlotinib with 300 mg ranitidine, an H2-receptor antagonist, decreased Erlotinib exposure [AUC] and maximum concentrations [Cmax] by 33% and 54%, respectively. Increasing the dose of Erlotinib when coadministered with such agents is not likely to compensate for this loss of exposure. However, when Erlotinib was dosed in a staggered manner 2 hours before or 10 hours after ranitidine 150 mg b.i.d., Erlotinib exposure [AUC] and maximum concentrations [Cmax] decreased only by 15% and 17%, respectively. The effect of antacids on the absorption of Erlotinib has not been investigated but absorption may be impaired, leading to lower plasma levels. In summary, the combination of Erlotinib with proton pump inhibitors should be avoided. If the use of antacids is considered necessary during treatment with Erlotinib, they should be taken at least 4 hours before or 2 hours after the daily dose of Erlotinib. If the use of ranitidine is considered, it should be used in a staggered manner; i.e. Erlotinib must be taken at least 2 hours before or 10 hours after ranitidine dosing.
Erlotinib and Gemcitabine: In a Phase Ib study, there were no significant effects of gemcitabine on the pharmacokinetics of Erlotinib nor were there significant effects of Erlotinib on the pharmacokinetics of gemcitabine.
Erlotinib and Carboplatin/Paclitaxel: Erlotinib increases platinum concentrations. In a clinical study, the concomitant use of Erlotinib with carboplatin and paclitaxel led to an increase of total platinum AUC0-48 of 10.6%. Although statistically significant, the magnitude of this difference is not considered to be relevant. In clinical practice, there may be other co-factors leading to an increased exposure to carboplatin like renal impairment. There were no significant effects of carboplatin or paclitaxel on the pharmacokinetics of Erlotinib.
Erlotinib and Capecitabine: Capecitabine may increase Erlotinib concentrations. When Erlotinib was given in combination with capecitabine, there was a statistically significant increase in Erlotinib AUC and a borderline increase in Cmax when compared with values observed in another study in which Erlotinib was given as single agent. There were no significant effects of Erlotinib on the pharmacokinetics of capecitabine.
Erlotinib and proteasome inhibitors: Due to the working mechanism, proteasome inhibitors including bortezomib may be expected to influence the effect of EGFR inhibitors including Erlotinib. Such influence is supported by limited clinical data and preclinical studies showing EGFR degradation through the proteasome.
Special precautions for disposal: No special requirements.
Incompatibilities: Not applicable.
Special precautions for storage: Do not store above 30°C.
L01EB02 - erlotinib ; Belongs to the class of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors. Used in the treatment of cancer.
Erlotinib Sandoz FC tab 150 mg
30's


 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image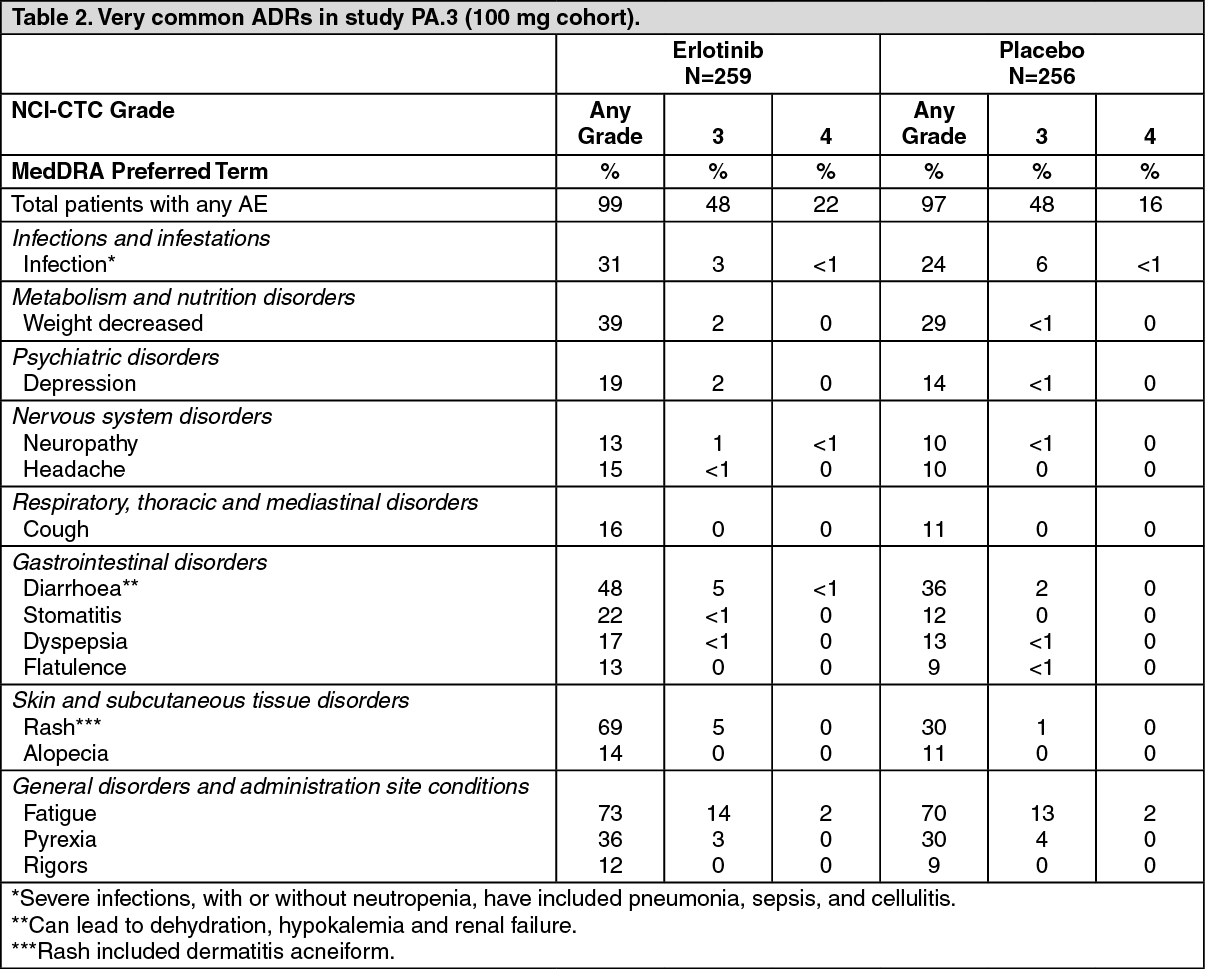 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image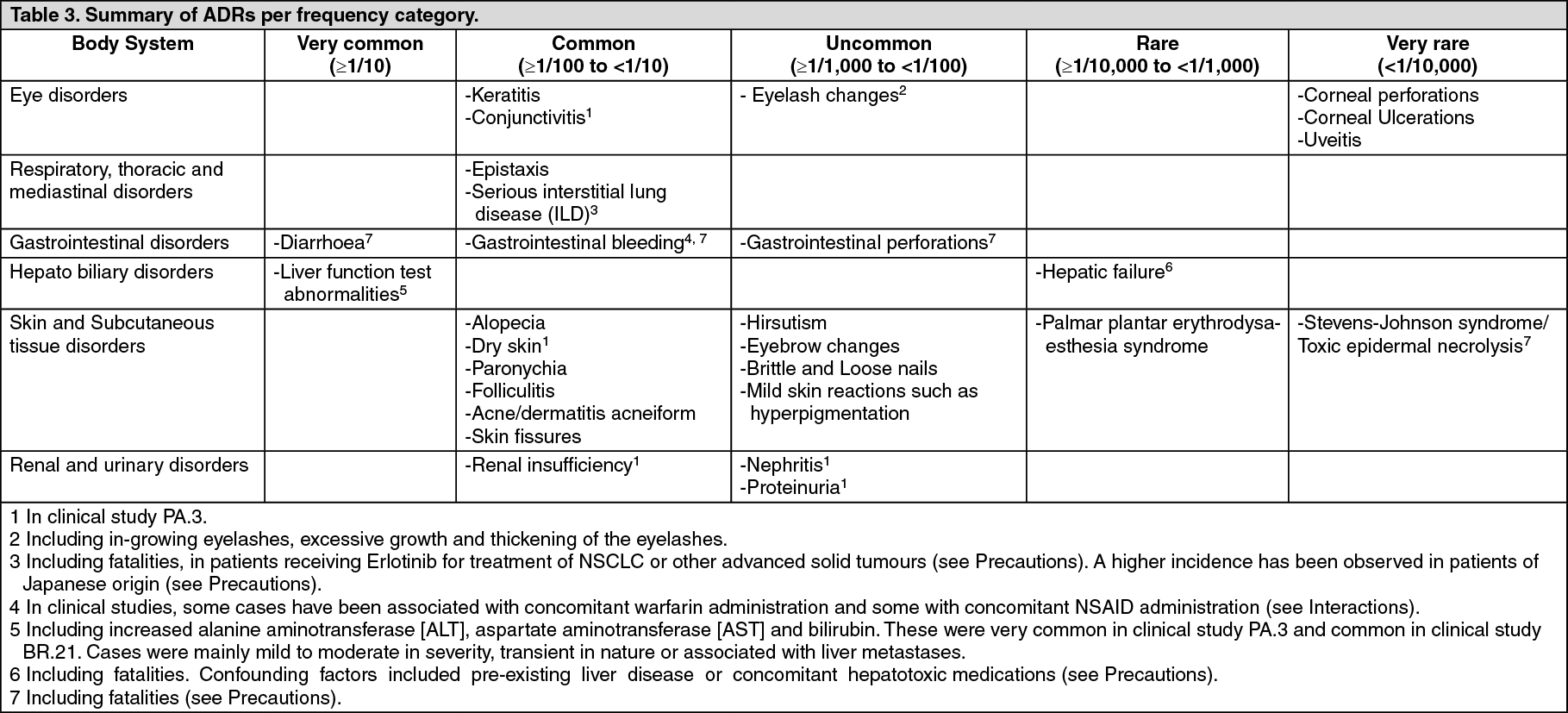 Click on icon to see table/diagram/image
Click on icon to see table/diagram/image
 Sign Out
Sign Out
